In May 2002, a 59-year-old male was admitted to our hospital because of extreme fatigue during the last week and elevated levels of urea and S-creatinine on biochemical tests. His past history included mild proteinuria since 5 years (last measurement 1,35 gr/day), cardiomyopathy since 7 years (he received verapamil treatment), chronic obstructive pulmonary disease (on inhalation therapy) since 5 years and partial deafness with tinnitus since 10 years. His family history was positive for renal disease: his mother had suffered from cardiomyopathy, deafness and chronic kidney disease (CKD) of unknown origin. On admission he did not report abnormalities of diuresis or fever.
Physical examination was regular except for reddish macular-papular skin lesions in the abdomen (which were reported since the age of 30 years) and blood pressure of 160/80 mmHg.
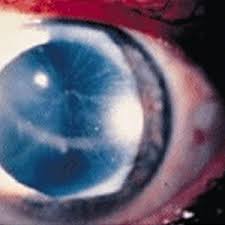
Laboratory examinations showed serum creatinine of 4,4 mg/dl (please provide SI units), BUN of 80.9 mg/ (please provide SI units),and moderate anaemia. In the urinary sediment 20-25 RBC/hmf (please explain) were found. 24-hour urinary protein excretion was 800 mg. A renal ultrasound revealed small, shrunken kidneys and a cardiac echo a thickening of the interventricular septum of 1,99 cm and a left ventricular outflow gradient of 67 mmHg. Ophthalmologic examination revealed tortuosity and ''sausage-like'' anomalies of the conjunctival and retinal vessels.
What is the diagnosis?
Answere:
Fabry disease . This diagnosis should be considered because of the symptoms and signs of cardiomyopathy, skin lesions, ocular abnormalities and family history of renal disease.
The clinical diagnosis of Fabry disease must be confirmed by assay of a-galactosidase activity in leukocytes or plasma and/or detection of GL3 deposition in tissue biopsies .
In our case a renal biopsy could not be performed. CT scan and MRI are no diagnostic tools for this case.
Since 2001, enzyme replacement therapy (ERT) is the only specific treatment for Fabry disease.Immunosuppressive drugs (corticosteroids and cyclosporine) have no effect on the disease. Gene therapy is a potential option for treatment of Fabry disease but is currently on development [3].Anticonvulsant drugs are used as symptomatic therapy of neuropathic pain due to Fabry disease .
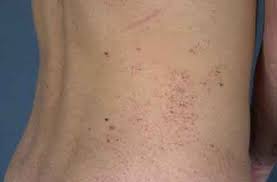

Physical examination was regular except for reddish macular-papular skin lesions in the abdomen (which were reported since the age of 30 years) and blood pressure of 160/80 mmHg.
Laboratory examinations showed serum creatinine of 4,4 mg/dl (please provide SI units), BUN of 80.9 mg/ (please provide SI units),and moderate anaemia. In the urinary sediment 20-25 RBC/hmf (please explain) were found. 24-hour urinary protein excretion was 800 mg. A renal ultrasound revealed small, shrunken kidneys and a cardiac echo a thickening of the interventricular septum of 1,99 cm and a left ventricular outflow gradient of 67 mmHg. Ophthalmologic examination revealed tortuosity and ''sausage-like'' anomalies of the conjunctival and retinal vessels.
What is the diagnosis?
Answere:
Fabry disease . This diagnosis should be considered because of the symptoms and signs of cardiomyopathy, skin lesions, ocular abnormalities and family history of renal disease.
The clinical diagnosis of Fabry disease must be confirmed by assay of a-galactosidase activity in leukocytes or plasma and/or detection of GL3 deposition in tissue biopsies .
In our case a renal biopsy could not be performed. CT scan and MRI are no diagnostic tools for this case.
Since 2001, enzyme replacement therapy (ERT) is the only specific treatment for Fabry disease.Immunosuppressive drugs (corticosteroids and cyclosporine) have no effect on the disease. Gene therapy is a potential option for treatment of Fabry disease but is currently on development [3].Anticonvulsant drugs are used as symptomatic therapy of neuropathic pain due to Fabry disease .
COMMENTS
Fabry disease is an X-linked lysosomal storage disorder caused by deficient or absent activity of the lysosomal enzyme α-galactosidase A. Insufficient activity of this enzyme causes the accumulation of glycosphingolipids with terminal α-galactosidic linkages, particularly globotriaosylceramide (GL-3), in various tissues and cell types . This process causes damage to endothelial, perithelial and smooth-muscle cells of the vascular system, glomerular and tubular cells of the kidney, myocardial cells and valvular fibrocytes, epithelial cells of the cornea and ganglion cells of the dorsal root and autonomic nervous system, as well as cortical and brain-stem structures. Therefore, the disease presents as a multi-system disorder, clinical features being typical but highly variable in affected individuals. Renal disease is one of the major causes of morbidity and mortality in Fabry patients. In classically affected males, the first renal manifestations can be observed as proteinuria, hematuria and lipiduria. In our patient, except for hematuria and proteinuria, Fabry disease was suspected also because of skin lesions and cardiac involvement. Even though ERT was started when the patient was on dialysis, there was a clinical improvement in his cardiac and renal function. Some studies refer to a more important clinical benefit for transplanted patients with Fabry disease . Different treatment approaches are currently on development. One of them implies the use of the active-site-specific chaperone 1-deoxygalactonojirimycin that acts facilitating folding of mutant GLA in the endoplasmic reticulum and increasing its lysosomal residual activity. Reduction of Gb3 deposits has been shown in lymphoblasts from Fabry patients with missense mutations and transgenic mouse model expressing a missense mutation GLA. Gene therapy has been also developed as a potential option for treatment of Fabry disease/


